Para que la Investigación Clínica continúe generando conocimiento es necesario asegurar que el entorno sigue siendo competitivo y cuenta con todos los factores adecuados para la ejecución de los ensayos. Se ha realizado un gran trabajo para adaptarnos a la nueva regulación, pero todavía queda camino por recorrer y sobre todo para superar pequeñas dificultades para finalizar el proceso de implementación de la regulación europea de ensayos clínicos.

La investigación clínica es una parte importante del progreso médico y el desarrollo de tratamientos eficaces para diversas patologías y afecciones médicas. sin embargo, la realización de ensayos clínicos conlleva una serie de desafíos, incluida la compleja regulación que desde 2022 rige dichos ensayos. La implementación de la regulación europea de ensayos clínicos 536/2014, ha marcado un hito significativo en la estandarización y la mejora de la calidad de la investigación clínica. En este artículo, exploraremos el impacto de esta regulación en la investigación clínica, sin olvidar la prioridad en 2024 para la implementación de esta regulación, que son las transiciones de los ensayos a la regulación europea.
La implementación de la Regulación Europea de Ensayos Clínicos ha promovido estándares más rigurosos y coherentes para la realización de ensayos clínicos en la UE. Esto ha llevado a una mayor coordinación entre los estados miembros involucrados en dichos ensayos, así como un único proceso de evaluación a través de todos los países. Además, la regulación ha simplificado los procesos administrativos y ha promovido la transparencia en la divulgación de resultados de ensayos clínicos, lo que beneficia tanto a los pacientes, como a la comunidad investigadora y a la población en general.
El 31 de enero de 2022 comenzó una implementación gradual de la regulación en las evaluaciones en los ensayos, haciéndose obligatoria para todos los ensayos clínicos iniciales a partir del 30 de enero de 2023 y siendo el 30 de enero de 2025, la fecha máxima para que todos los ensayos de la UE operen bajo el sistema CTIS y, por lo tanto, bajo la regulación dejando atrás la antigua directiva.
A pesar de las muchas ventajas de la regulación europea, su implementación no está exenta de desafíos y obstáculos. Uno de los principales desafíos está siendo la necesidad de adaptar los sistemas y procesos existentes para todos los stakeholders para cumplir con los nuevos requisitos regulatorios. Esto ha supuesto una importante inversión tanto de tiempo como de recursos por parte de todos los agentes implicados, pero con un dinamismo necesario para la actualización de los procesos. En la actualidad, tenemos procesos, documentos y guías adaptados que están en continuo cambio, consolidándose de acuerdo con cómo progresa la implementación.
Tanto las agencias reguladoras, como los comités de ética, promotores, centros de investigación etc., estamos atravesando por diferentes fases de aprendizaje y consolidación de los procesos. La eficiencia en la evaluación y homogénea gestión de los ensayos es el principal objetivo que estamos superando día a día en basándonos en los aprendizajes a lo largo de las experiencias actuales.
Las experiencias operacionales, recientemente compartidas por EMA, arrojan números totalmente alentadores a nivel de implantación (gráfico 1) podemos observar el total de los ensayos clínicos presentados a evaluación, 3698, un número elevado que transmite el claro enfoque de los promotores para la rápida asimilación de la regulación.
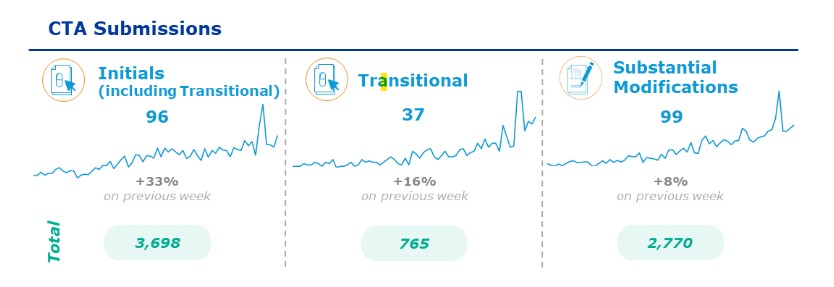
*CTIS newsflash – 9 February 2024
Los datos en azul en el gráfico 1 representan los ensayos clínicos presentados a evaluación desde el 30 de enero al 5 de febrero de 2024. Estos son elevados en el periodo reducido de muestra, dato que aumentará gradualmente a lo largo del año, lo que permitirá que todos los implicados aumenten su conocimiento de CTIS y por lo tanto el dominio de los procesos.
Otro de los datos en gráfico, representan el número de ensayos que han pasado a ser legislados bajo la regulación; (hasta el momento, 765), siendo este el objetivo principal a corto plazo de todos los promotores, ya sean comerciales o no. El periodo de transición finalizará el 30 de enero de 2025 por lo tanto para todos aquellos ensayos clínicos que estén activos a partir de esa fecha será necesario asegurar que operan bajo el Reglamento. Nuestra reciente experiencia en el año 2016, con el cambio de la Directiva 2001 al Real Decreto 1090/2015, fue del todo positiva al permitir AEMPS que los ensayos finalizarán bajo la regulación derogada. Esto habría sido un gran acierto si se hubiera podido llevar a cabo de la misma manera con el reglamento.
Desde EMA y junto con los estados miembros se ha hecho un gran esfuerzo en establecer normas claras y sobre todo ágiles, para facilitar dicha transición. Así contamos con una guía que establece un procedimiento acelerado y simplificado, en el que se definen 22 días de evaluación, para el cambio de todos los ensayos que estén activos más allá de enero del 2025. Sin embargo, aunque la norma esté escrita, siempre hay grises y tanto los promotores como resto de stakeholders seguimos anticipando un año difícil debido a las barreras que identificamos para la transición. Entre otras se podrían señalar las siguientes:
- Necesidad de encontrar el momento adecuado en la vida del ensayo para poder pasar los ensayos a la nueva regulación. Será necesario hacer un análisis exhaustivo de los documentos aprobados bajo el antiguo Real Decreto, (o bajo directiva) y homogeneizar documentos a lo largo de los estados miembros implicados, para que todos los países involucrados cuenten con las mismas versiones aprobadas. El hecho de encontrar un momento de “silencio” en el ensayo y homogeneizar los documentos de éste, requiere una gran planificación y coordinación entre los países, siendo este el punto de partida para la transición.
- Falta de definición en la guía de EMA sobre el seguimiento del procedimiento acelerado de los estados miembros. Es decir; cabe la posibilidad de que estados miembros soliciten documentación adicional, que redundará en tiempo dedicado para la recopilación de ésta, dedicación por tanto de recursos del promotor y del equipo investigador y un gran impacto en los tiempos de transición, siendo este un gran impacto en el proceso y tiempos de aprobación.
- Se anticipa “un cuello de botella” en la preparación y evaluación de los ensayos a pasar a la regulación. Todos los agentes implicados están orientando su enfoque a la rápida preparación y transición de estos. Los temores son que algún ensayo no esté autorizado cuando finalice el periodo máximo de transición. Ante este escenario, los agentes implicados y en concreto los promotores, tenemos algunas preguntas importantes sin respuesta; ¿Cuáles serán las implicaciones si no da tiempo a obtener la autorización de algún ensayo que ha realizado la transición? ¿Habrá alguna información oficial a promotores para estas situaciones? Y lo que es más importante ¿Cómo afectará al paciente en el ensayo, teniendo en cuenta que puede ser que esté con tratamientos activos? Todas estas cuestiones están pendientes de definir y son consideradas una gran preocupación, de ahí que se demande más concreción en la información.
El procedimiento acelerado ha atenuado claramente el momento más inquietante en el proceso de transición. Ha sido necesario experimentar con las solicitudes presentadas, para ir poco a poco refinando el proceso. Lo que sigue siendo complejo es contar siempre con la misma postura de todos los estados miembros implicados. A través de los países de la EU nos encontramos con diversos tiempos de evaluación y requerimientos aplicables a la evaluación de una transición, lo que hace que se gestione de manera diferente muchas de las solicitudes de transición. Nuestra esperanza es que cuanto antes los países vayan adquiriendo experiencia en el proceso de evaluación coordinado y se observe el beneficio lo antes posible.
Con respecto al objetivo fundamental de este año 2024 y hasta el fin de enero de 2025, no quiero dejar de identificar, el siguiente paso, que no es obligatorio, una vez que el ensayo requiera una modificación relevante. Será en este momento cuando se actualizarán todos aquellos documentos que, haciendo uso del proceso acelerado, no se actualizaron con los requerimientos de la regulación, pero que, en el momento de esta modificación sí son requeridos. Es decir, nos enfrentamos a una modificación sustancial que se progresará como un ensayo inicial, y que por lo tanto requiere planificación, coordinación, generación de documentos y sobre todo comunicación para que la evaluación comience y se produzca cuanto antes.
Como hemos visto a lo largo del artículo, se trata de una tarea ardua implementar la regulación europea de ensayos clínicos. Hasta el momento, el resumen, tras ver los ensayos presentados y conocer cómo se ha implementado en todos los países, es muy alentador y estimulante. Habría que reconocer el gran esfuerzo realizado por todas las partes en tiempos muy reducidos, aunque todavía hay que seguir trabajando la redefinición de alguno de esos procesos y colaboraciones con otros países. Sería recomendable que las revisiones siempre fueran orientadas a ganar en eficiencia en la evaluación, con la intención de reducirla en el tiempo y evitar repeticiones de preguntas en los momentos de alegaciones a los protocolos.
Es responsabilidad de todos trabajar en continua dedicación y con un enfoque claro para alcanzar el cumplimiento de los objetivos principales de la regulación. De esta forma podremos ver cómo las eficiencias planificadas se plasmarán, en el desarrollo de nuevos tratamientos a lo largo del territorio. Es importante tener en mente nuestra región y sobre todo la gran contribución que hace la UE a la investigación, para que recuperemos el liderazgo hasta hace poco mantenido en torno a la investigación clínica. Centrar toda nuestra atención y acción en el paciente y las futuras estrategias terapéuticas de las que se beneficiará, es lo único que justifica nuestra dedicación constante para que la regulación europea de ensayos clínicos sea ejemplarizante en su implementación.











